فهرست مطالب
مقاله جامع، تخصصی و علمی در زمینه اتوفاژی (Autophagy)، تهیه شده توسط دکتر غلامرضا ابری برای مجله اینترنتی «فارماوب»
اتوفاژی (Autophagy): مکانیسمهای مولکولی، نقش در بیماریها و استراتژیهای دارویی درمانی
در این مقاله تخصصی از فارماوب، مکانیسمهای مولکولی اتوفاژی، نقش دوگانه آن در سرطان و بیماریهای عصبی، و آخرین استراتژیهای دارویی شامل مهارکنندههای کلروکین و فعالکنندههای AMPK را بررسی میکنیم.
کلمات کلیدی (Keywords):
اتوفاژی, Autophagy, mTOR, AMPK, مهارکننده اتوفاژی, کلروکین, هیدروکسیکلروکین, درمان سرطان, بیماریهای عصبی, فارماوب, اتوفاژی و ایمونوتراپی.
اتوفاژی: از مکانیسمهای بنیادی تا استراتژیهای درمانی نوین در پزشکی دقیق
نویسنده: دکتر غلامرضا ابری / تیم تحریریه فارماوب)
منبع: مجله داروسازی در اینترنت (فارماوب)
مقدمه:
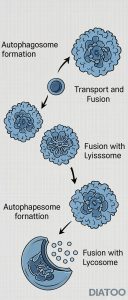
اتوفاژی (Autophagy) به فارسی خودخواری سلولی، از واژه یونانی به معنای «خودخواری»، فرآیندی تکاملی محافظتشده و حیاتی در سلولهای یوکاریوتی است که برای حفظ هموستازی سلولی و زنده ماندن در شرایط استرس ضروری است. این فرآیند شامل تجزیه و بازیافت اجزای سیتوپلاسمی، پروتئینها و اورگانل های آسیبدیده است. در سال ۲۰۱۶، جایزه نوبل فیزیولوژی و پزشکی به یوشینوری اوهسومی برای کشف ژنها و مکانیسمهای اتوفاژی تعلق گرفت، که اهمیت این فرآیند را در سلامت و بیماری برجسته ساخت.
در این مقاله، به بررسی مکانیسمهای مولکولی اتوفاژی، نقش آن در بیماریهای مختلف، و استراتژیهای دارویی درمانی میپردازیم.
۱. مکانیسمهای مولکولی و تنظیم اتوفاژی
خواخواری سلولی فرآیندی چندمرحلهای است که با دقت توسط مسیرهای سیگنالینگ پیچیده تنظیم میشود. سه نوع اصلی اتوفاژی شناخته شده است: ماکرواتوفاژی (Macroautophagy)، مایکرواتوفاژی (Microautophagy)، و اتوفاژی میانجیشده توسط کورپورون (Chaperone-Mediated Autophagy, CMA). در اینجا، تمرکز اصلی بر ماکرواتوفاژی است که به طور ساده به عنوان اتوفاژی شناخته میشود.
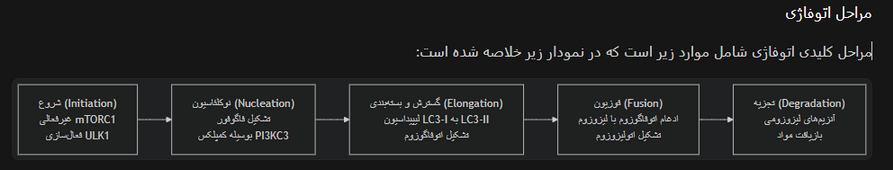
مراحل اتوفاژی
مراحل کلیدی اتوفاژی شامل موارد زیر است که در نمودار زیر خلاصه شده است:
مسیرهای سیگنالینگ تنظیمکننده
دو مسیر اصلی سیگنالینگ که نقش کلیدی در تنظیم اتوفاژی دارند، mTOR (mammalian Target of Rapamycin) و AMPK (AMP-activated protein kinase) هستند.
- mTORC1 به عنوان مهارکننده اصلی: در شرایط غنی از مواد مغذی، mTORC1 فعال است و با فسفریلاسیون ULK1 در سایت Ser757، از تعامل ULK1 با AMPK جلوگیری کرده و اتوفاژی را مهار میکند.
- AMPK به عنوان فعالکننده اصلی: در شرایط کمبود انرژی (افزایش نسبت AMP/ATP)، AMPK فعال میشود. AMPK دو راه اصلی برای فعالسازی اتوفاژی دارد: (۱) مستقیماً ULK1 را در سایتهای Ser317 و Ser777 فسفریله میکند که منجر به فعالسازی آن میشود؛ (۲) با فسفریلاسیون TSC2 و Raptor، فعالیت mTORC1 را مهار کرده و به طور غیرمستقیم منجر به فعالسازی ULK1 میشود.
📖 جزئیات بیشتر درباره پروتئینهای کلیدی ATG
خانواده پروتئینهای ATG (AuTophaGy-related) نقش اصلی را در اجرای مراحل اتوفاژی بر عهده دارند. برخی از مهمترین آنها عبارتند از:
– ULK1/ATG1: کیناز آغازین که مسیر سیگنالینگ را به ماشینهای اتوفاژی متصل میکند.
– Beclin-1/ATG6: بخشی از کمپلکس PI3KC3 است که برای نوکلئاسیون فاگوفور ضروری است.
– ATG5-ATG12-ATG16L1: کمپلکس که به عنوان E3-like لیگاز عمل کرده و به لیپیداسیون LC3 کمک میکند.
– LC3/ATG8: پروتئینهای مستعد لیپیداسیون که به غشای فاگوفور متصل شده و برای بستهبندی و حمل بار ضروری هستند. فرم LC3-II به عنوان مارکر اتوفاگوزومها استفاده میشود.
– p62/SQSTM1: آداپتوری که پروتئینهای ubiquitinated را به LC3-II متصل کرده و selective autophagy را ممکن میسازد.
۲. نقش دوگانه اتوفاژی در بیماریها
اتوفاژی نقشی دوگانه و context-dependent در بسیاری از بیماریها ایفا میکند، که آن را به یک هدف درمانی چالشبرانگیز اما جذاب تبدیل کرده است.
۲.۱. اتوفاژی و سرطان: شمشیر دو لبه
اتوفاژی در سرطان نقشی پیچیده و مرحلهوابسته دارد:
- نقش سرکوبگر تومور (مراحل اولیه): در سلولهای سالم، اتوفاژی با حفظ یکپارچگی ژنومی، جلوگیری از تجمع پروتئینهای سمی و کاهش التهاب مزمن، از بدخیمی جلوگیری میکند. کاهش فعالیت اتوفاژی با افزایش خطر ابتلا به برخی سرطانها مرتبط است.
- نقش تسهیلکننده تومور (مراحل پیشرفته): در تومورهای تثبیتشده، سلولهای سرطانی از اتوفاژی برای بقا در محیطهای hypoxic و کمنوتیریت، مقاومت به درمانهای (مانند شیمیدرمانی و پرتودرمانی) و متاستاز استفاده میکنند. در این مرحله، مهار اتوفاژی میتواند به کاهش تومور و افزایش حساسیت به درمان کمک کند.
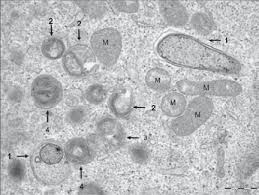
۲.۲. اتوفاژی و بیماریهای عصبی
اختلال در اتوفاژی با تجمع پروتئینهای misfolded (مانند β-amyloid و Tau در آلزایمر، α-synuclein در پارکینسون، و huntingtin در بیماری هانتینگتون) مرتبط است.
در این بیماریها:
- اتوفاژی با حذف پروتئینهای سمی و اورگانلهای آسیبدیده (مانند میتوکندریها)، عملکرد نورونها را حفظ میکند.
- با این حال، اتوفاژی بیش از حد یا dysregulated نیز ممکن است به مرگ نورونی کمک کند (“autophagy paradox”). بنابراین، هدف قرار دادن اتوفاژی در بیماریهای عصبی نیازمند تنظیم دقیق و context-specific است.
مدولاسیون اتوفاژی به یک استراتژی جذاب برای درمان طیف وسیعی از بیماریها تبدیل شده است. دو رویکرد اصلی شامل مهار و فعالسازی اتوفاژی است.
۳.۱. مهارکنندههای اتوفاژی: تمرکز بر سرطان
مهار اتوفاژی به طور گستردهای به عنوان یک استراتژی adjutant در درمان سرطان، به خصوص برای غلبه بر مقاومت به درمان، در حال بررسی است.
- کلروکین (CQ) و هیدروکسیکلروکین (HCQ): تنها داروهای مهارکننده اتوفاژی هستند که برای استفاده بالینی تأیید شدهاند (برای malaria و بیماریهای خودایمنی). این داروها با افزایش pH لیزوزومی، از فوزیون اتوفاگوزوم با لیزوزوم و تجزیه محتوای آن جلوگیری میکنند.
- کاربردهای بالینی: دهها کارآزمایی بالینی فاز I/II اثر HCQ را به عنوان تکدارو یا در ترکیب با شیمیدرمانی، هدفدرمانی (مانند مهارکنندههای mTOR یا BRAF)، و ایمونوتراپی در انواع سرطانها (مانند پانکراس، پروستات، کلیه، و گلیوبلاستوما) بررسی کردهاند.
- نتایج نشان دادهاند که HCQ به طور کلی ایمن است اما به عنوان تکدارو اثریت محدودی دارد. ترکیب آن با سایر درمانها امیدوارکنندهتر است.
- چالشها: HCQ یک مهارکننده نسبتاً ضعیف است و برای مهار کامل اتوفاژی در تومورهای انسانی، نیاز به دوزهای بالا دارد که ممکن است با سمیت (مانند رتینوپاتی و کاردیوتوکسیسیتی) همراه باشد.
- همچنین، مانیتورینگ pharmacodynamic اتوفاژی در بیماران چالشبرانگیز است.
🔬 نمونهای از کارآزمایی بالینی: HCQ در سرطان پانکراس
در یک کارآزمایی فاز II در بیماران مبتلا به سرطان پانکراس متاستاتیک پیشدرمانشده، HCQ (400 یا ۶۰۰ mg دو بار در روز) تجویز شد. نتیجه نشان داد که تنها ۱۰٪ بیماران پس از دو ماه بدون پیشرفت بیماری ماندند. median PFS و OS به ترتیب ۴۶.۵ و ۶۹ روز بود. عوارض درجه ۳/۴ شامل لنفوپنی و افزایش ALT بود. این مطالعه نشان داد که HCQ به عنوان تکدارو اثریت محدودی دارد اما ترکیب آن با سایر درمانها ممکن است مفیدتر باشد.
- مهارکنندههای جدیدتر و قدرتمندتر: برای غلبه بر محدودیتهای CQ/HCQ، چندین کلاس جدید از مهارکنندههای اتوفاژی در حال توسعه هستند. این شامل مشتقات Lys01 (مانند Lys05) که قدرتمندتر هستند و مهارکنندههای dual-target (مانند مهارکنندههای همزمان اتوفاژی و REV-ERB) است. این عوامل در پیشبالینی نشان دادهاند که مهار اتوفاژی و رشد تومور را به طور مؤثری تقویت میکنند.
۳.۲. فعالکنندههای اتوفاژی: پتانسیل در بیماریهای عصبی و متابولیک
فعالسازی اتوفاژی ممکن است برای حذف پروتئینهای سمی در بیماریهای عصبی و بهبود متابولیسم در بیماریهایی مثل دیابت مفید باشد.
- مهارکننده های mTOR: داروهایی مانند رپامایسین (Sirolimus) و مشتقات آن (Everolimus, Temsirolimus) با مهار mTORC1، اتوفاژی را القا میکنند.
- این داروها به عنوان immunosuppressants و در درمان برخی سرطانها (مانند سرطان کلیه) استفاده میشوند. اثرات محافظتی عصبی آنها در مدلهای حیوانی بیماریهایی مثل آلزایمر و پارکینسون نشان داده شده است، اما استفاده بالینی برای این indications هنوز در مراحل اولیه است.
- AMPK فعالکنندهها: داروهایی مانند متفورمین (Metformin)، AICAR و natural compounds مانند رزوراترول (Resveratrol) با فعالسازی AMPK و مهار mTOR، اتوفاژی را القا میکنند.
- متفورمین، داروی دیابت، نشان داده است که میتواند خطر ابتلا به برخی سرطانها و شواهدی از اثرات مفید در بیماریهای عصبی دارد، که ممکن است تا حدی از طریق القای اتوفاژی میانجی شود.
- سایر فعالکنندهها: داروهایی مانند Trehalose (قند دیساکارید) و Spermidine نیز نشان دادهاند که میتوانند از طریق مسیرهای وابسته به TFEB (عامل اصلی transcriptional برای اتوفاژی و لیزوزوژنز) اتوفاژی را فعال کنند.
۳.۳. ترکیب با ایمونوتراپی: یک مرز جدید
شواهد فزایندهای نشان میدهد که اتوفاژی با ایمونوتراپی، به خصوص با مهارکنندههای checkpoint (مانند anti-PD-1/PD-L1) تعامل دارد
- اتوفاژی میتواند ارائه آنتیژن (antigen presentation) توسط سلولهای سرطانی و سلولهای دندریتیک را تنظیم کند.
- مهار خودخواری سلولی ممکن است به افزایش پاسخ ایمنی ضد تومور و غلبه بر مقاومت به ایمونوتراپی کمک کند. کارآزماییهای بالینی در حال بررسی ترکیب HCQ با ایمونوتراپی در انواع سرطانها هستند.
۴. چالشها و آینده مداخلات دارویی بر اساس اتوفاژی
اگرچه اتوفاژی هدفی جذاب است، اما چندین چالش مهم برای ترجمه موفقیتآمیز به کلینیک وجود دارد:
- دوگانگی نقش: همانطور که بحث شد، اتوفاژی میتواند نقش محافظتی یا پیشرونده داشته باشد. این امر نیاز به درک دقیق context هر بیماری و مرحله آن برای تصمیمگیری در مورد فعالسازی یا مهار اتوفاژی را ایجاد میکند.
- lack of biomarkers: برای انتخاب بیمارانی که بیشتر از مداخلات خودخواری سلولی بهرهمند میشوند و برای مانیتورینگ پاسخ درمانی، نیاز به biomarkers قابل اعتماد است. LC3-II در بافت تومور یا سلولهای خونی محیطی به عنوان یک candidate مطالعه شده است، اما نتایج در بیماران ناسازگار بوده است
- سمیت و selectivity: مهارکنندگان فعلی مثل HCQ selectivity پایینی دارند و دوزهای بالا نیاز دارند که میتواند عوارض داشته باشد. توسعه مهارکنندههای قدرتمندتر و selectiveتر یک اولویت است.
- combination therapies: ترکیب مدولاتورهای خودخواری سلولی با سایر درمانها (شیمیدرمانی، هدفدرمانی، ایمونوتراپی) به نظر میرسد استراتژی موثرتری باشد، اما تعیین دوزهای بهینه و برنامههای زمانی برای جلوگیری از سمیت افزایش (synergistic toxicity) ضروری است.
آینده این زمینه در توسعه:
- مدولاتورهای selectiveتر که به طور خاص اجزای ماشین اتوفاژی را هدف قرار میدهند.
- پزشکی دقیق (Precision Medicine): استفاده از genomic و molecular profiling برای پیشبینی اینکه کدام تومورها به “اتوفاژی addiction” وابسته هستند و به مهار آن پاسخ میدهند.
- درمانهای ترکیبی هوشمند: طراحی رژیمهای ترکیبی بر اساس in-depth درگیری مسیرهای سیگنالینگ در تومورهای خاص.
نتیجهگیری
این مرحله عجیب فرآیندی بنیادی و حیاتی برای سلامت سلولی است که نقش پیچیدهای در طیف وسیعی از بیماریها ایفا میکند. درک عمیقتر مکانیسمهای مولکولی و نقش context-dependent آن، پایه و اساس توسعه استراتژیهای درمانی جدید است. اگرچه مهارکنندههای فعلی مانند HCQ محدودیتهایی دارند، اما مسیر روشنی برای توسعه نسل جدیدی از مدولاتورهای اتوفاژی با قدرت و selectivity بیشتر وجود دارد. ترکیب این عوامل با سایر درمانها، به خصوص ایمونوتراپی، پتانسیل تغییر پارادایم در درمان سرطان و بیماریهای دیگر را دارد. آینده پژوهش و داروسازی در زمینه اتوفاژی بسیار امیدوارکننده است و ممکن است به درمانهای انقلابی برای بیماریهایی که تا کنون درمان مؤثری نداشتند منجر شود.
منابع (References):
- Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell. 2011;145(1):22-35.
- Kim J, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132-141.
- Amaravadi RK, et al. Targeting autophagy in cancer: moving from bench to bedside. Nat Rev Clin Oncol. 2019;16(7):407-420.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27-42.
- Mauthe M, et al. Chloroquine inhibits autophagic flux by limiting autophagosome-lysosome fusion. Autophagy. 2018;14(8):1435-1455.
- Ravikumar B, et al. Rapamycin-mediated inhibition of mTORC1 induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. J Neurosci. 2004;24(35):7805-7812.
سلب مسئولیت: محتوای این مقاله صرفاً جهت اطلاعرسانی علمی است و جایگزین توصیه پزشک متخصص یا داروساز نیست. استفاده از هر دارو، به خصوص off-label، باید تحت نظارت دقیق پزشک انجام شود.